21-羟化酶缺乏症是一种常染色体隐性遗传疾病,由肾上腺合成皮质醇所需的一种酶缺乏引起。21-羟化酶缺乏是先天性肾上腺增生的最常见原因,糖皮质激素和盐皮质激素替代治疗是其主要治疗方法。
CYP21A2基因突变导致 21-羟化酶缺乏,CYP21A2基因提供了制造称为 21-羟化酶的酶。这种酶存在于肾上腺中,它在产生称为皮质醇和醛固酮的激素中发挥作用。
皮质醇具有多种功能,例如维持血糖水平、保护身体免受压力和抑制炎症;醛固酮有时被称为盐潴留激素,因为它调节肾脏保留的盐量,盐分的滞留会影响体内的液体水平和血压。
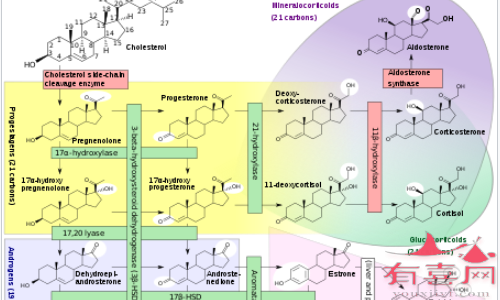
21-羟化酶缺乏症是由 21-羟化酶缺乏引起的,当缺乏 21-羟化酶时,通常用于形成皮质醇和醛固酮的物质反而会在肾上腺中积聚并转化为雄激素。雄激素的过量产生会导致 21-羟化酶缺乏症患者的性发育异常;缺乏醛固酮产生会导致患有这种情况的盐浪费形式的人的盐分流失。
功能性 21-羟化酶的量决定了疾病的严重程度,耗盐型个体具有导致完全无功能的酶的CYP21A2突变。
患有这种情况的简单男性化类型的人具有CYP21A2基因突变,可以产生低水平的功能性酶,患有这种疾病的非经典类型的个体具有CYP21A2突变,导致产生的酶量减少,但比其他任何一种类型的酶都多。
21羟化酶缺乏症遗传学
P450c21 酶(即:21-羟化酶)的CYP21A2基因位于编码主要人类组织相容性基因座(HLA)的基因HLA B和HLA DR的 6p21.3中。
CYP21A2与无功能的假基因CYP21A1P配对,CYP21A2异常等位基因的评分已被记录,大部分来自CYP21A2和CYP21A1P同源区域的重组。
21-羟化酶缺乏可能是由大约30 Kb的大缺失引起的,其中不仅包括CYP21A2基因的大部分区域,还包括CYP21A1P假基因的所有C4B基因和区域。CYP21A1P 假基因和 C4B 基因的重复通常与非经典 21-羟化酶缺乏症有关。由于CYP21A2基因与CYP21A1P假基因同源性高,且位点复杂,分子水平研究较为困难。
各种等位基因残留酶活性的差异解释了疾病的不同严重程度,所有形式的 21-羟化酶 CAH 的遗传都是常染色体隐性遗传。
受任何形式疾病影响的人都有两个异常等位基因,父母双方通常都是杂合子或携带者。当父母双方都携带异常等位基因时,每个孩子有 25% 的机会患上这种疾病,有 50% 的机会像父母一样成为携带者,有 25% 的机会拥有两个正常基因。
现在可以通过测量ACTH刺激后的17α-羟孕酮升高来测试杂合性,或者最近通过直接基因测序来测试。
迄今为止,已鉴定出CYP21A2基因中的 200 多种致病变异,如果这些变异中至少有两种以复合杂合子的形式存在,则会导致 21-羟化酶缺乏症。
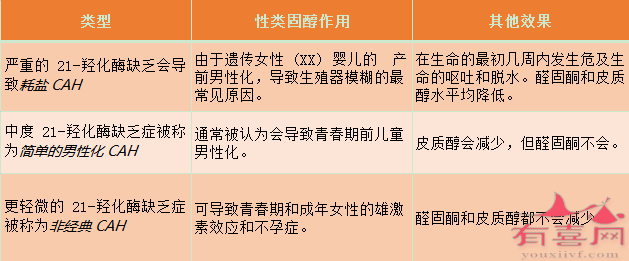
21-羟化酶缺乏症分为三种类型,其中两种类型是经典形式,称为盐浪费和简单的男性化类型,第三种类型称为非经典类型。盐浪费型最严重,单纯男性化型较轻,非经典型最轻。
1.经典的 21-羟化酶缺乏症
盐浪费型
大约 75% 的典型 21-羟化酶缺乏症患者属于盐浪费型,这是该病最严重形式,通常与导致酶没有活性的大基因缺失或内含子突变有关。
简单的男性化
大约 25% 的典型 21-羟化酶缺乏患者表现为简单的男性化。简单的男性化形式最常见的原因是点突变导致氨基酸取代,导致低但可检测的酶活性导致足够的醛固酮分泌,但皮质醇水平降低。
2.非经典 21-羟化酶缺乏症
非经典型更为常见,具有非经典形式的女性可能是具有经典突变和变异等位基因的复合杂合子或具有两个变异等位基因的杂合子,允许正常酶活性的20%至60%。
复合杂合子女性的表型较轻,临床表现各不相同。女性可以出现在任何年龄,但通常不小于 6 个月。杂合子女性可能有轻微的生化异常,但没有临床上重要的内分泌疾病。
1.经典的 21-羟化酶缺乏症
失盐型
皮质醇和醛固酮两种合成途径的CYP21都受累。新生儿患者除了具有单纯男性化型的临床表现外,还有水盐失衡的表现;如拒乳、呕吐、脱水、休克,如得不到及时救治,死亡率甚高。失盐型患者大多数在出生后1~5周发病,6周以后发病者极少。

单纯男性化型
只有皮质醇合成途径的CYP21受累,醛固酮合成途径正常。
①女性患者外生殖器有不同程度男性化(从阴蒂肥大到大阴唇融合,形成部分性阴茎尿道)。严重的病例一般有生殖窦存留(阴道和尿道只有一个开口),卵巢、子宫和输卵管存在,附睾和输精管缺如。雄激素水平增高的另一个作用是使身体直线生长加速,身材比同龄儿童高,骨龄提前,ACTH产生过多可引起皮肤色素沉着。
②男性患者表现为非促性腺激素释放激素(GnRH)依赖性性早熟,阴茎增大,阴毛生长,但是睾丸和促性腺激素仍停留在青春期前水平。皮肤色素沉着和身体直线生长加速与女性患者表现相同。
2.非经典 21-羟化酶缺乏症
①无症状型:又称隐性CYP21缺乏症,在经典型CYP21缺乏症家系中,有些成员无男性化表现,但是血清CYP21催化反应步骤的前体类固醇17-OHP水平增高。
②迟发型:出生时外生殖器正常,没有男性化改变。而在青春期前出现多毛、痤疮、身体直线生长加速和骨龄提前。
诊断
1.经典型患者的 17-羟基孕酮会非常高(通常大于 1000 ng/dL)
2.高钾血症、低钠血症、低醛固酮和高血浆肾素活性 (PRA),尤其是 PRA 与醛固酮的比率,是盐皮质激素合成受损的标志
3.应进行 ACTH 刺激测试以评估肾上腺功能并区分各种潜在的酶缺陷。给予 0.25 mg cosyntropin(一种合成的促肾上腺皮质激素)对肾上腺提供药理刺激,最大限度地提高激素分泌。
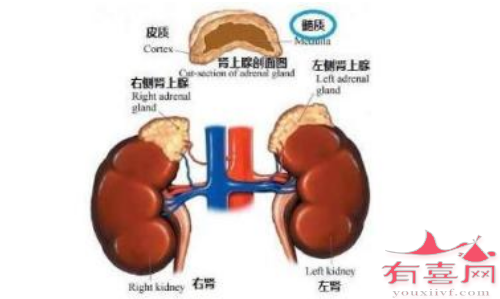
4.完整的肾上腺情况,包括 17-羟基孕酮 (17-OHP)、皮质醇、脱氧皮质酮、11-脱氧皮质醇、17-羟基孕烯醇酮、脱氢表雄酮 (DHEA) 和雄烯二酮的测量,应在施用促肾上腺皮质激素之前和 60 分钟后立即获得。
5.染色体核型分析
6.盆腔超声检查以检查子宫或相关的肾脏异常、骨龄检查、肾上腺CT等相应检查
治疗
21-羟化酶缺乏症治疗方式包括药物及手术治疗,患者需要终身间歇性治疗。
1.药物治疗
糖皮质激素:氢化可的松是基础用药,需要终生的替代治疗。应分别按照患者尚在生长中和已达到成年身高情况制定方案。未停止生长者,建议用氢化可的松替代。达到成年身高后,可以给半衰期相对长的制剂,如泼尼松或地塞米松。在应激状态和疾病时需对糖皮质激素的剂量进行调整。
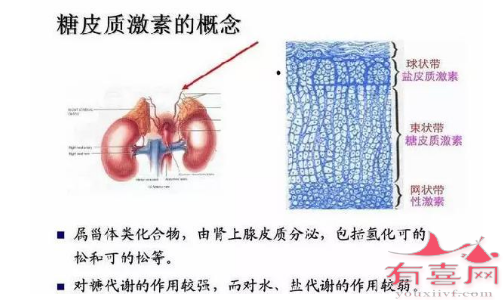
盐皮质激素:21-羟化酶缺乏症失盐型患者在糖皮质激素基础上补充盐皮质激素,可以减少糖皮质激素的总量及长期不良反应。
生长激素和促性腺激素释放激素类似物:对于性早熟严重、骨龄超前明显、预测成年身高损失较多者,可考虑生长激素治疗。对于已经发生中枢性性早熟的患者,可使用促性腺激素释放激素类似物。
手术治疗
生殖器不明确的婴儿可能需要进行矫正手术,阴蒂成形术可在治疗病情稳定后、1岁前进行,阴道成形术可在成年后进行。
对多数得到恰当治疗的女性,可有正常的月经并可能怀孕,只是自然怀孕的几率较低。
原因包括:不合适的阴道入口导致不满意的性生活、阴道的性交疼痛、因雄激素水平高而导致卵巢功能紊乱、以及对性别认知与选择性伴侣的性心理行为。长期排卵停止、孕激素升高和异位子宫内膜着床也常被认为是生育力下降的原因。
而男性生育力下降的主要原因是睾丸肾上腺残余瘤,其被认为起源于异位的肾上腺组织。此外,低促性腺素性功能减退症可因LH分泌受到肾上腺雄激素及其芳香化产物分泌过多的抑制作用导致。

综上,21羟化酶缺乏症在得到恰当的治疗后是有机会生育的,只是相比正常人生育几率较低。且21羟化酶缺乏症属于比较常见的遗传疾病,自然生育后代有患上该病的风险。
因此,21羟化酶缺乏症患者想要生育先要进行详细的生育咨询与遗传咨询,看是否有自然生育的能力。针对不孕不育患者,建议进行三代试管婴儿,三代试管婴儿不仅能够解决不孕不育问题,还能够筛除遗传致病基因,生育健康的宝宝。
以上是关于“21羟化酶缺乏症可以生孩子吗,有哪些临床表现”的相关内容,希望对您有所帮助。