●基本信息
【疾病名称】珠蛋白生成障碍性贫血
【别名】地中海贫血、海洋性贫血
【外文名】thalassemia
【就诊科室】血液科
【常见病因】珠蛋白链合成不足或缺如导致
【常见症状】贫血,肝脾大进行性加重,黄疸,发育不良等
●概述
珠蛋白生成障碍性贫血又称地中海贫血,海洋性贫血症,是遗传性血液疾病,会造成血红蛋白合成障碍,其症状可依不同分型而有所不同,程度可能从无症状到严重。
通常珠蛋白生成障碍性贫血伴随典型的贫血症状,即红血球细胞水准低下。贫血可导致疲累感与肤色苍白,也可同时造成骨骼疾病、脾脏肿大、黄疸、深色尿以及儿童成长迟缓等症状。
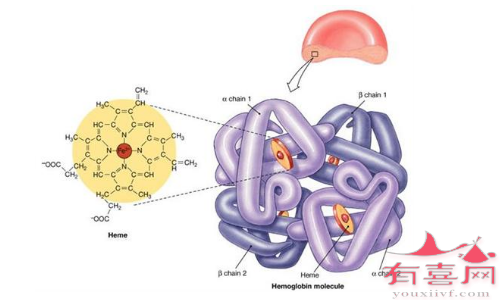
●病因
1.α珠蛋白生成障碍性能超群贫血正常人自父母双方各继承2个α珠蛋白基因(αα/αα),合成足够的α珠蛋白链。若自父母继承一个或一个以上有缺陷的α基因,则导致α珠蛋白生成障碍性贫血,临床表现的严重程度取决于异常α基因的数目。
若一个基因异常(α-/αα)患者无血液学异常表现;若2个α基因异常(α-/α-或-/αα),红细胞呈小细胞低色素性改变,无显著性溶血和贫血;若3个α基因异常(-/α-),有代偿性溶血性贫血表现,如血红蛋白H病;若4个α基因异常,无α珠蛋白链生成,导致胎儿水肿综合征(HbBarts)。
2.β珠蛋白生成障碍性贫血正常人自父母双方各继承一个正常β珠蛋白基因,合成正常量的β珠蛋白链。若自父母继承了异常β基因,则导致本病。
自父母一方继承一个异常β基因,从另一方继承一个正常β基因,患者则为杂合子,即β珠蛋白生成障碍性贫血,有约半量β链合成,病情减轻。
若自父母双方各继承一个异常β基因,则患者为纯合子,即β0珠蛋白生成障碍性贫血,没有或极少β链生成,病情严重。
●发病机制
1.α珠蛋白生成障碍性贫血
引起本病的基因异常多数是基因缺失,α基因缺失数目多少与α珠蛋白链缺乏程度及临床表现严重性平行。但少数患者并无α基因缺失,而是由于α基因发生点突变或数个碱基缺失,影响了RNA加工、mRNA翻译或导致合成的α珠蛋白链不稳定,最终引起α珠蛋白链缺乏。
当α珠蛋白链缺乏时,没有足够的α珠蛋白链与β链配对形成HbA,剩余的β链形成HbH(β4),而β亲氧力过高,不宜运输氧,且不稳定,发生沉淀,形成包涵体,引起溶血。当α链完全缺失时,胎儿期无HbF(α2γ2)形成,仅有HbBarts(γ4),而γ4有极高亲氧力,不能向组织释放足够氧,以致胎儿缺氧而死。
胎儿水肿综合征:胎儿父母均为正常α基因和α0基因的杂合子,即父母双方均为(--/αα),胎儿不幸继承了父母双方的α0基因,即4个α基因均缺失,无α链生成。正常胎儿HbF(α2γ2)缺失,而γ链自行聚合形成HbBarts(γ4)。HbBarts的氧亲和力高,向组织释放氧很少,导致胎儿窒息死亡。
本病胎儿大多在妊娠30~40周时成为死胎而流产,或早产后数小时而死亡。
血红蛋白H病:患者父母中一方是α0珠蛋白生成障碍性贫血性状(--/αα),另一方是α珠蛋白生成障碍性贫血性状(αα/-α),患者自父母双方继承了异常α基因(-/-α)导致了血红蛋白H病。
本病患者仅能生成少量α珠蛋白链,α/β链合成比例约为0.3(正常比例应为1.0),由于α链不足,无α链配对的β链自行聚合成β4四聚体,即HbH。
HbH不仅氧亲和力高,向组织释放氧少,而且不稳定,易在红细胞内发生沉淀形成HbH包涵体,导致红细胞在脾内破坏。
2.β珠蛋白生成障碍性贫血
本病的分子病理相当复杂,已知有50种以上的β基因突变可导致本病。这些基因突变大致可划分为5类:
①基因片段缺失,导致β链合成缺失;②转录突变,单一碱基的点突变发生在β基因转录调控区内,降低β基因的转录,导致β链合成不足;③加工突变,即点突变发生在影响mRNA加工形成的区域,导致β链合成缺失或不足;④终止密码突变,点突变发生在翻译终止密码上,形成异常mRNA,导致β链合成缺失;⑤移码突变,一个或数个(非3个或3的倍数)碱基的缺失或插入,使β基因碱基排列发生框架移位,导致β链合成缺失。由于β链的不足或缺失,未能与β链配对的α链是不稳定的,会发生沉淀,在红细胞内形成α链包涵体,导致红细胞在骨髓内或脾脏内破坏。
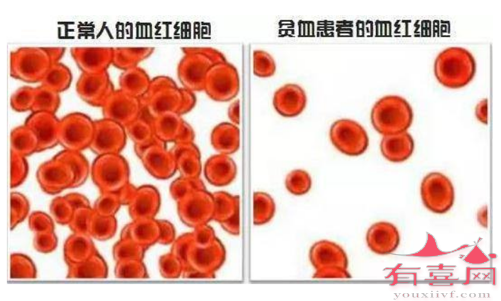
本病的HbA(α2β2)减少而HbF(α2γ2)、HbA2(α2δ2)增多,HbF增多的机制可能是HbF的红细胞(称F细胞)寿命较长缘故,HbA2增多的机制一是HbA相对减少,二是β基因突变使相邻的δ基因表达增高缘故。
●临床表现
1.α球蛋白生成障碍性贫血
出生时即可贫血,临床表现为轻~中度的慢性贫血,伴有黄疸、肝脾肿大;继发感染、服用氧化剂药物可加重HbH的不稳定性而促发溶血;合并妊娠可加重贫血。
患者的发育一般不受影响,骨骼改变也不明显。
2.纯合子(β0)球蛋白生成障碍性贫血
患儿出生时正常,出生数月后,HbF逐渐被HbA(α2β2)替代,患儿出现贫血,呈进行性加重,可有发热、纳差、腹泻、黄疸、肝、脾逐渐肿大。3~4岁时,表现生长发育迟缓,精神萎靡,面无表情,体弱无力。

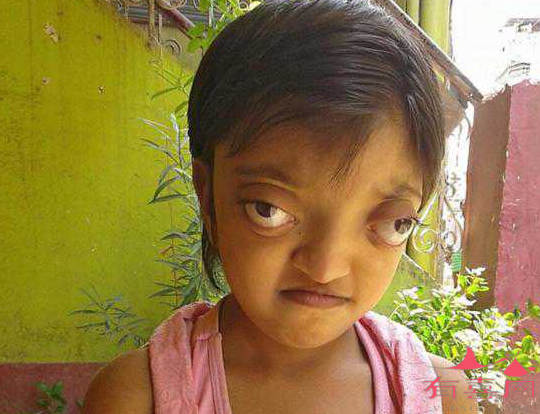
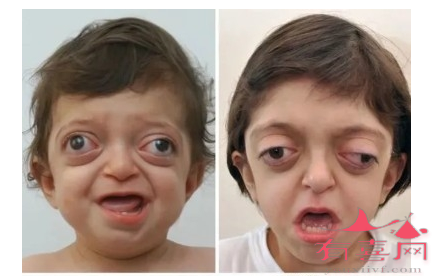
骨髓造血代偿性增生使骨髓腔变宽骨皮质变薄,导致患儿额部、顶部隆起,头颅增大,面颊隆起,鼻梁塌陷,上颌及牙齿前突,形成特殊面容,患者发生下肢慢性溃疡。
发病愈早,症状愈重。
3.杂合子(β)珠蛋白生成障碍性贫血
此类患者又称静止型或微型β珠蛋白生成障碍性贫血,因为大多数患者无贫血或其他症状,多在普查、家系调查或合并其他疾病进行检查时发现。查血或可发现少数靶形红细胞、红细胞渗透脆性轻度降低、HbA2轻度增多。
少数患者可有贫血,尤其当合并妊娠或继发感染时,表现为轻度至中度贫血,可出现黄疸、轻度脾大。
根据病情轻重的不同,分为以下3型。
1.重型
出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型的表现是臀状头,长骨可骨折。
骨骼改变是骨髓造血功能亢进、骨髓腔变宽、皮质变薄所致。
少数患者在肋骨及脊椎之间发生胸腔肿块,亦可见胆石症、下肢溃疡。
2.中间型
轻度至中度贫血,患者大多可存活至成年。
3.轻型
轻度贫血或无症状,一般在调查家族史时发现。
●诊断
1.β珠蛋白生成障碍性贫血
(1)重型外周血象呈小细胞低色素性贫血,红细胞大小不等,中央浅染区扩大,出现异形、靶形、碎片红细胞和有核红细胞、点彩红细胞、嗜多染性红细胞、豪-周氏小体等;网织红细胞正常或增高。骨髓象呈红细胞系统增生明显活跃,以中、晚幼红细胞占多数,成熟红细胞改变与外周血相同;红细胞渗透脆性明显减低。HbF含量明显增高,大多>0.40,这是诊断重型β地贫的重要依据。颅骨X线片可见颅骨内外板变薄,板障增宽,在骨皮质间出现垂直短发样骨刺。
(2)轻型成熟红细胞有轻度形态改变,红细胞渗透脆胜正常或减低,血红蛋白电泳显示HbA2含量增高(0.035~0.060),这是本型的特点。HbF含量正常。
(3)中间型外周血象和骨髓象的改变如重型,红细胞渗透脆性减低,HbF含量为0.40~0.80,HbA2含量正常或增高。
2.α珠蛋白生成障碍性贫血
(1)静止型红细胞形态正常,出生时脐带血中HbBart's含量为0.01~0.02,但3个月后即消失。
(2)轻型红细胞形态有轻度改变,如大小不等、中央浅染、异形等;红纽胞渗透脆性降低;变性珠蛋白小体阳性;HbA2和HbF含量正常或稍低。患儿脐血HbBart's含量为0.034~0.140,于生后6个月时完全消失。
(3)中间型外周血象和骨髓象的改变类似重型β地贫;红细胞渗透脆性减低;变性珠蛋白小体阳性;HbA2及HbF含量正常。出生时血液中含有约0.25HbBart's及少量HbH;随年龄增长,HbH逐渐取代HbBart's,其含量为0.024~0.44。包涵体生成试验阳性。
●治疗
轻型地贫无需特殊治疗,中间型和重型地贫应采取下列一种或数种方法给予治疗。输血和去铁治疗,在目前仍是重要治疗方法之一。
1.一般治疗
注意休息和营养,积极预防感染。适当补充叶酸和维生素B12。
2.红细胞输注
输血是治疗本病的主要措施,最好输入洗涤红细胞,以避免输血反应。少量输注法仅适用于中间型α和β地贫,不主张用于重型β地贫。对于重型β地贫应从早期开始给予中、高量输血,以使患儿生长发育接近正常和防止骨骼病变。其方法是:先反复输注浓缩红细胞,使患儿血红蛋白含量达120~150g/L;然后每隔2~4周输注浓缩红细胞10~15ml/kg,使血红蛋白含量维持在90~105g/L以上。但本法容易导致含铁血黄素沉着症,故应同时给予铁螯合剂治疗。
3.铁螯合剂
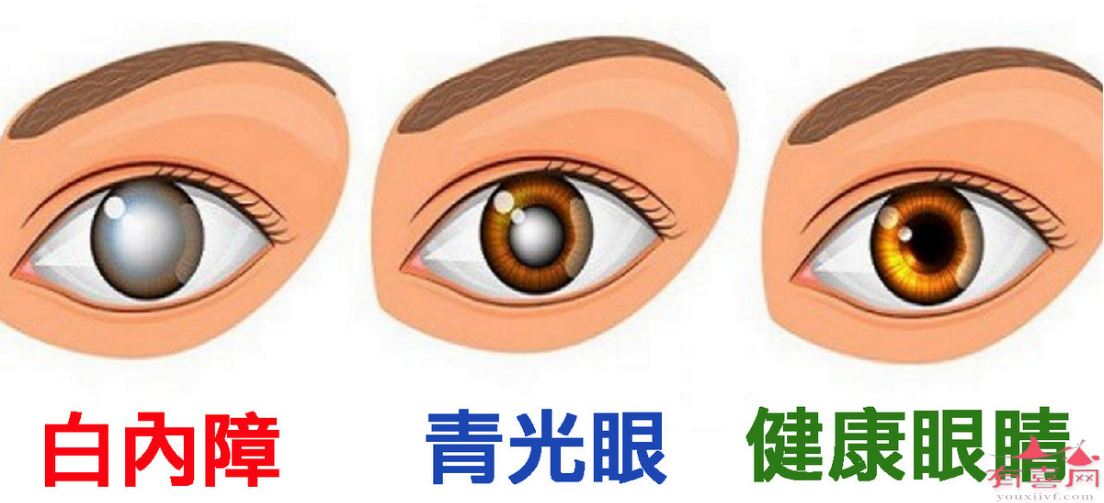
常用去铁胺,可以增加铁从尿液和粪便排出,但不能阻止胃肠道对铁的吸收。通常在规则输注红细胞1年或10~20单位后进行铁负荷评估,如有铁超负荷则开始应用铁螯合剂。去铁胺,每晚1次连续皮下注射12小时,或加入等渗葡萄糖液中静滴8~12小时;每周5~7天,长期应用。或加入红细胞悬液中缓慢输注。去铁胺副作用不大,偶见过敏反应,长期使角偶可致白内障和长骨发育障碍,剂量过大可引起视力和听觉减退。维生素C与螯合剂联合应用可加强去铁胺从尿中排铁的作用。
4.脾切除
脾切除对血红蛋白H病和中间型β地贫的疗效较好,对重型β地贫效果差。脾切除可致免疫功能减弱,应在5~6岁以后施行并严格掌握适应证。
5.造血干细胞移植异基因
造血干细胞移植是目前能根治重型β地贫的方法。如有HLA相配的造血干细胞供者,应作为治疗重型β地贫的首选方法。
6.基因活化治疗
应用化学药物可增加γ基因表达或减少α基因表达,以改善β地贫的症状,已用于临床的药物有羟(经)基脲、5-氮杂胞苷(5~AZC)、阿糖胞苷、马利兰、异烟肼等,目前正在研究中。
【注:】具体采用何种治疗方式,如何展开治疗,需根据患者自身情况考虑,以上内容仅供了解。
在婚配方面,应向患者提出医学建议防止下一代发生纯合子β0珠蛋白生成障碍性贫血。在夫妻均为杂合子β珠蛋白生成障碍性贫血,应对胎儿进行产前基因诊断,避免纯合子β0珠蛋白生成障碍性贫血患儿的出生。
鉴于本病缺少根治疗法,预后不良,故应提倡阳性家族史者进行婚前检查和胎儿产前基因诊断,避免纯合子β珠蛋白生成障碍性贫血患儿出生。
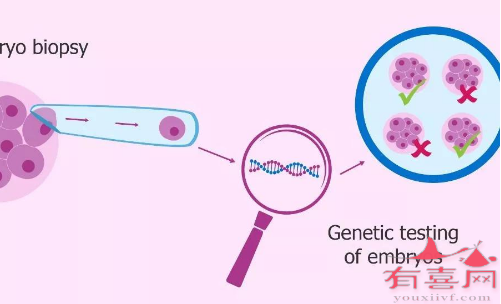
目前第三代试管婴儿的基因筛查技术能够有效预防珠蛋白生成障碍性贫血继续遗传给后代。