●基本信息
【疾病名称】先天性肾上腺皮质增生症
【别名】肾上腺生殖器综合征/肾上腺性变态征
【外文名】Congenitaladrenalhyperplasia(CAH)
【就诊科室】儿科
【常见病因】皮质激素合成酶的先天缺陷
【常见症状】男性化,失盐表现(食欲差,呕吐,嗜睡和体重增加缓慢)
●概述
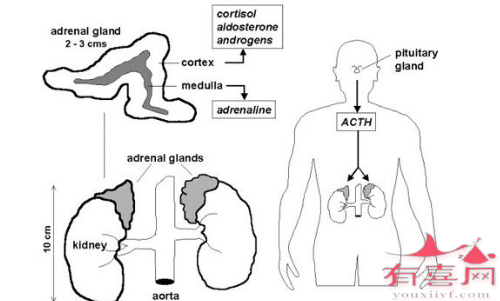
先天性肾上腺增生症(CAH)一组罕见的、影响肾上腺的常染色体隐性遗传疾病,肾上腺是肾脏上方的一对核桃大小的器官,肾上腺产生重要的激素,包括:
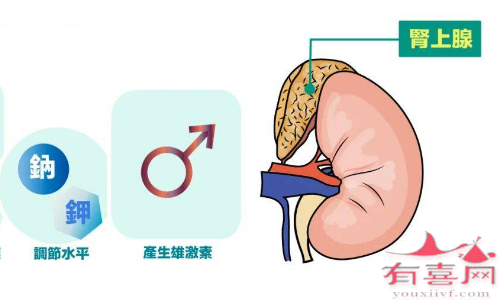
皮质醇,调节身体对疾病或压力的反应
盐皮质激素,如醛固酮,可调节钠和钾水平
雄激素,例如睾酮,是男性性激素
在患有CAH的人中,遗传问题导致缺乏制造这些激素所需的一种酶。
虽然没有治愈方法,但只要治疗得当,大多数患有先天性肾上腺增生症的人都能过上正常的生活。
先天性肾上腺增生有两种主要类型:
一、经典CAH。这种形式较为罕见,通常在婴儿期被发现。大约75%患有经典CAH的人有所谓的失盐形式,而25%的人有所谓的单纯男性化形式。
二、非经典CAH。这种形式更温和、更常见,可能要到童年或成年早期才会显现出来。
●经典CAH
失盐形式
在75%的严重酶缺乏的病例中,醛固酮分泌不足会导致盐分流失、不能茁壮成长,以及潜在的致命的低血容量和休克。失盐型CAH的漏诊与新生儿早期发病和死亡的风险增加有关。
单纯男性化形式
新生女婴CAH的主要特征是外生殖器的发育异常,有不同程度的男性化现象。根据临床实践指南,对于发现有双侧不可触及的性腺的新生儿,应考虑进行CAH评估。如果不能鉴别和治疗男性化的CAH,男孩和女孩都可能在出生后快速生长并出现男性化。
●非经典CAH
除了在婴儿期诊断的失盐型和单纯男性化CAH外,还存在一种轻度或非经典型,其特点是出生后有不同程度的雄性激素过多,但有时无症状。
非经典型症状可能在儿童后期才会凸显,并可能导致生长加速、性早熟、痤疮和继发性多囊卵巢综合征。在成年男性中,早期秃头和不育可能是该病的症状。
非经典型的特点是皮质醇合成的轻度亚临床损伤;血清皮质醇浓度通常正常。
通常来讲,经典的CAH是更严重的形式,如果不被发现和治疗,可能导致肾上腺危象和死亡;非经典CAH较温和,可能出现也可能不出现症状。
●其他罕见类型形式
11-β羟化酶缺乏症、 17a-羟化酶缺乏症、3-β-羟基类固醇脱氢酶缺乏症、先天性类脂肾上腺增生和p450氧化还原酶缺乏症,它们都表现出不同的症状。
●经典CAH症状
失盐CAH患者也缺乏醛固酮激素,导致无法调节保留身体盐和水的水平,这可能会导致失水过多而脱水、低循环血容量和异常低血压甚至休克。
如果不进行治疗,这种严重的CAH会因肾上腺危象导致严重的虚弱、呕吐、腹泻和循环衰竭。
单纯男性化CAH患者存在异常增大的肾上腺,会产生过量的雄激素,受该疾病影响的女性性发育异常。由于21-羟化酶缺乏而患有严重或典型男性化CAH的女性,很可能有不明确或不典型的外生殖器,尽管她们在遗传上是女性并且具有正常的内部生殖器官,而患有这种类型CAH的男性不会有模棱两可的生殖器。
如果在早期没有得到诊断和治疗,不论男女都可能出现其他症状,例如青春期提前、身体快速生长和过早完成生长导致身材矮小。
●非经典CAH症状
非经典CAH患者不会危及生命,症状通常在儿童后期出现,儿童后期的症状可能包括过早的体毛或痤疮发展。
在青春期女性中,最常见的问题包括面部或体毛过多、月经不调和脓疱性痤疮。无论男女都有正常的生殖器,一小部分非经典CAH人群具有低生育能力。非经典CAH患者可能不需要治疗也不影响正常生活。
●罕见的CAH形式
11-β羟化酶缺乏症患者可以避免出现与肾上腺危象相关的症状,尽管他们会受到其他症状的影响,例如由于水潴留和女性生殖器模棱两可导致的高血压。
17a-羟化酶缺乏导致男性外生殖器模糊,女性缺乏青春期发育或月经周期(闭经)。
3-β-羟基类固醇脱氢酶缺乏会导致男性和女性的生殖器不明确。在两种性别中,它都会导致盐分浪费。
先天性类脂性肾上腺增生可能因肾上腺危象而导致早期死亡。男性有模棱两可的生殖器,男性和女性如果存活下来,都可能无法生育。
P450氧化还原酶缺乏症的体征和症状可能类似于21-羟化酶缺乏症、17-羟化酶缺乏症或两种酶缺乏症的组合。有些病例与称为Antley-Bixler综合征的骨骼疾病有关。
CAH最常见的原因是缺乏21-羟化酶,负责21-羟化酶的基因中的不同突变导致酶的不同水平,产生一系列影响。
CYP21A2基因的缺失和突变是所有21-羟化酶缺乏型CAH病例的原因。
CYP11B1、CYP17A1、HSD3B2、CYP11A1、STAR和CYPOR基因的突变分别导致11-羟化酶、17-羟化酶、3-β-羟基类固醇脱氢酶缺乏、类脂肾上腺增生和PORD钙。
所有形式的CAH均以常染色体隐性遗传模式遗传(如下图)。
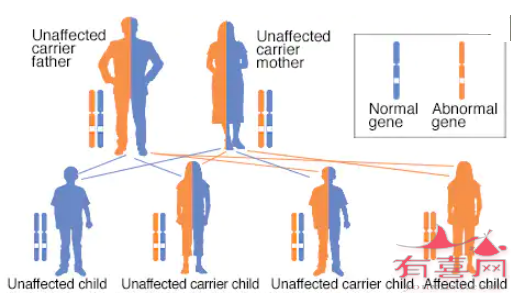
只有从父母双方各遗传一个突变基因,才会患上常染色体隐性遗传疾病。这些疾病通常遗传自两名携带者,携带者的健康很少受到影响,但他们拥有这种疾病的一个突变基因(隐性基因)和一个正常基因(显性基因)。
因此两名携带者生育带有两个正常基因的正常孩子的几率为25%(图左),生育同为携带者的健康孩子的几率为50%(图中),生育带有两个隐性基因的患病孩子的几率为25%(图右)。
●检查方法
1.骨龄检测
骨龄检测是通过对手腕、手和骨盆中生长板的X光片,可用于确定婴儿或儿童是否具有与其年龄相称的高级骨骼发育。可以帮助医生监测非典型先天性肾上腺增生症儿童的生长情况,也可用于监测患有经典病症的儿童的治疗。

2.ACTH1-24兴奋试验
对于经典型21-羟化酶缺陷症患者,根据临床表现和基础17-OHP(17-羟孕酮),一般可以明确诊断。血清17-OHP基础值不能提供足够的诊断依据时,有必要进行ACTH(促肾上腺皮质激素)1-24兴奋试验。一般而言60分钟时17-OHP水平在10ng/ml以上考虑非经典型21羟化酶缺陷症的诊断。每个实验室都应根据21羟化酶缺陷症杂合子携带者和正常人确定出自己的诊断标准。
3.性染色体检查
女性细胞核染色质为阳性,男性则为阴性,女性染色体计数性染色体为XX,男性则为XY,可确定其真正性别。
4.失盐的检查
PRA(血浆肾素活性)值升高,特别是PRA与24小时尿醛固酮比值增加标志着醛固酮合成障碍。在循环血中ACTH,17-0HP和孕酮水平高,但醛固酮水平正常的病例中这些指标也会升高,这样没有很好控制的单纯男性化患者的生化表现会与失盐型混淆。盐皮质激素治疗可以抑制这些患者的肾上腺,有助于二者的鉴别。理想状态下,血浆和尿醛固酮水平应该与PRA和钠平衡相关,从而有助于对临床类型的准确判断。在分析肾素水平的意义时,必须注意新生儿正常值高于年龄较大的儿童。
5.B型超声检查
先天性肾上腺皮质增生女性假两性畸形的内生殖器正常,B超和经插管X线造影能显示子宫和输卵管。B超、CT、MRI有助于鉴别肾上腺增生或肿瘤,先天性增生为双侧肾上腺对等增大,而肿瘤多为单侧孤立肿块,可有钙化,因出血和坏死可形成液化腔。
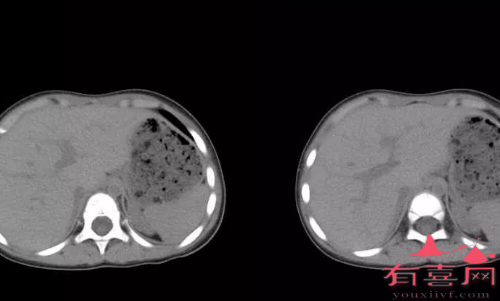
6.其他
女性肾上腺皮质增生假两性畸形者,用尿道镜检查尿生殖窦,可见阴道开口于子宫颈,若家族中有21-羟化酶缺乏者,可采用聚合酶链式反应(PCR)、羊膜细胞HLA分型和DNA进行分析。
●治疗方式
1.及时纠正水、电解质紊乱
主要针对失盐型患儿,静脉补液可用生理盐水,有代谢性酸中毒则用0.45%氯化钠和碳酸氢钠溶液。忌用含钾溶液。重症失盐型需静脉滴注氢化可的松,若低钠和脱水不易纠正,则可肌肉注射醋酸脱氧皮质酮(DOCA)或口服氟氢可的松,脱水纠正后,糖皮质激素改为口服,并长期维持,同时口服氯化钠。其量可根据病情适当调整。
2.药物治疗
经典CAH患者一生都需要每天服药。,如果患者停止服药,症状会再次出现。
(1)糖皮质激素糖皮质激素治疗一方面可补偿肾上腺分泌皮质醇的不足,一方面可抑制过多的ACTH释放,从而减轻雄激素的过度产生,故可改善男性化、性早熟等症状,保证患儿正常的生长发育过程。

(2)盐皮质激素盐皮质激素可协同糖皮质激素的作用,使ACTH的分泌进一步减少。可口服氟氢可的松,症状改善后,逐渐减量,停药。因长期应用可引起高血压。0.1mg氟氢可的松相当于1.5mg氢化可的松,应将其量计算于皮质醇的用量中,以免皮质醇过量。
在皮质激素治疗的过程中,应注意监测血17-羟孕酮或尿17-酮类固醇,失盐型还应该监测血钾、钠、氯等,因为身体在生命的不同时期会产生不同量的皮质醇,因此有时患者的药物剂量可能过高或过低,需要及时调整。
服用过多的药物来替代皮质醇会导致库欣综合征的症状。这些包括:
体重增加、增长放缓、皮肤上的妊娠纹、圆脸、高血压、骨质流失、高血糖
如果出现这些症状,请务必告知医生,以便调整药物剂量。
3.手术治疗
男性患儿无需进行手术治疗,女性患儿可能需要手术,例如,如果生殖器的变化影响了尿流,则需要进行手术。
女性两性畸形患儿宜6个月~1岁时进行阴蒂部分切除术或矫形术,但具体手术治疗时间父母应与孩子主治医生多交流,以确定最佳治疗时机。
1.产前诊断
CAH在妊娠约14周时通过绒毛膜绒毛取样(CVS)进行产前诊断,或在约20周后通过羊膜穿刺术进行产前诊断。
胎儿生殖器官在妊娠约9周时开始形成,胎儿雄激素过多会导致女性胎儿的生殖器男性化,为了防止受经典CAH影响的女性胎儿的生殖器模糊不清,在妊娠9周前开始给母亲服用地塞米松。
绒毛膜绒毛取样(CVS)是一种产前检查,用于检测出生缺陷、遗传疾病和怀孕期间的其他问题。在测试期间,从附着在子宫壁上的胎盘中取出一小部分细胞(称为绒毛膜绒毛)。
绒毛膜绒毛是由受精卵形成的胎盘的微小部分,因此它们具有与婴儿相同的基因。
2.三代试管婴儿
CAH患者想要避免将该病继续遗传给后代,生育健康宝宝,可以借助三代试管婴儿技术,在胚胎移植入母体之前,将异常基因胚胎排除,选取健康胚胎进行移植孕育。
目前不论国内外,三代试管婴儿技术都是相当纯熟的了,而CAH作为一种常染色体隐性遗传的单基因疾病,符合三代试管PGD技术的适应症,能够从根本上解决遗传问题,生育健康宝宝。